The Sutton and Iwase laboratories have a joint mission to uncover chromatin regulatory mechanisms underlying Smith-Magenis Syndrome (SMS). We hope that a complete understanding of these mechanisms will reveal specific alterations in the brain that give rise to core symptoms of SMS and, importantly, guide development of novel treatment approaches. Our groups bring complementary expertise to this mission – the Sutton laboratory has spent years studying molecular mechanisms that control information transfer across synaptic connections in the brain, while the Iwase laboratory has uncovered key ways in which neuronal activity modifies chromatin to control gene expression and neuronal function.

Our recent work has focused on the SMS causative gene Retinoic Acid Induced-1 (RAI1) and its role in regulating changes in neuronal gene expression that typically allow neurons to fine-tune synaptic connections to buffer destabilizing activity in neural circuits. We have found that RAI1 is critical for preventing inappropriate activation of these gene expression programs when neural circuit activity is stable, and we are interested in whether loss of this key checkpoint contributes to altered brain development in SMS. Our ongoing work uses a combination of cutting-edge technologies to identify RAI1 target genes in neurons, the degree to which these RAI1 targets are unique to human vs. rodent neurons, and how these target genes act to fine-tune synaptic function. We are excited by the possibility that these studies will identify new RAI1-regulated genes that can be targeted for future treatment of SMS.
2022-2023
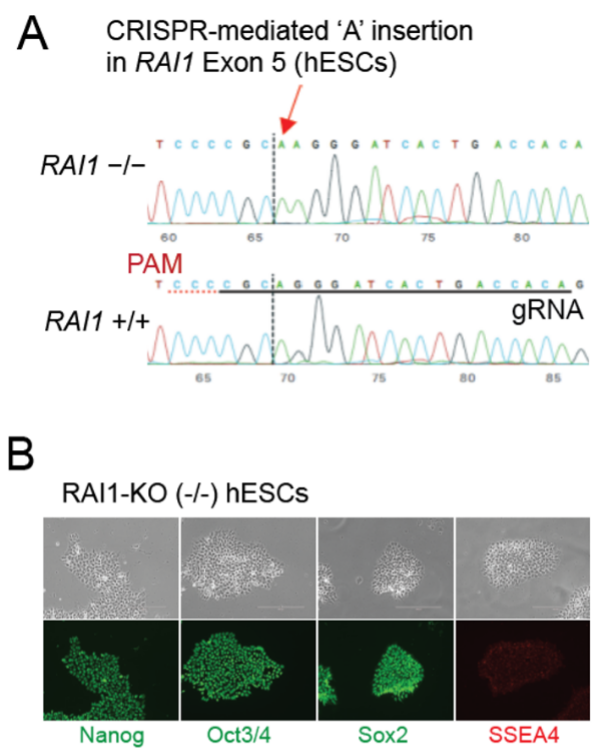
With generous support from the SMS Research Foundation, we used CRISPR-mediated gene editing to generate human embryonic stem cells (hESCs) with one or both copies of RAI1 deleted. We then differentiated these hESCs into cortical neurons while performing mRNA sequencing at key developmental stages and electrophysiological recordings of synaptic function. These experiments revealed a number of important findings: 1) Robust gene dysregulation was observed in RAI-deficient cells at early developmental time-points, suggesting a key role for this gene in early brain maturation; 2) Loss of RAI1 altered the developmental trajectory of differentiating neurons and produced accelerated neural development; 3) As we observed previously in rodent neurons, hESC-derived neurons deficient in RAI1 exhibited stronger synaptic connections under basal conditions and altered plasticity of those connections relative to control neurons.

2024-
With continued support from the SMS Research Foundation, we will use single-cell RNA sequencing (scRNAseq) to extend our previous work by profiling RAI-regulated genes in different human cell types as they differentiate into mature neurons. These experiments will address: 1) whether loss of RAI1 alters early differentiation into neural precursor cells and other cell lineages; 2) whether RAI1 gene dysregulation is similar in different cell types or is more pronounced in cells of the nervous system; 3) in mature neurons, what are the RAI1 target genes in different excitatory and inhibitory neuronal subtypes? We expect these studies to reveal key genes and molecular pathways dysregulated by the loss of RAI1 that we can target for future treatments for SMS.
The project is from University of Michigan. The two researchers are:
Michael Sutton, PhD
Professor of Molecular and Integrative Physiology
Shigeki Iwase PhD
Associate Professor of Human Genetics
